gen-pairs is for pair generation. The default is ‘no’,
i.e. get 1-4 parameters from the pairtypes list. When
parameters are not present in the list, stop with a fatal error. Setting
‘yes’ generates 1-4 parameters that are not present in the pair list
from normal Lennard-Jones parameters using fudgeLJ
fudgeLJ is the factor by which to multiply
Lennard-Jones 1-4 interactions, default 1
fudgeQQ is the factor by which to multiply
electrostatic 1-4 interactions, default 1
NN is the power for the repulsion term in a 6-NN potential (with
nonbonded-type Lennard-Jones only), starting with GROMACS version 4.5,
grompp
also reads and applies NN, for values not equal to 12 tabulated
interaction functions are used (in older version you would have to use
user tabulated interactions).
Note that gen-pairs,
fudgeLJ, fudgeQQ, and NN are optional.
fudgeLJ is only used when generate pairs is set to ‘yes’,
and fudgeQQ is always used. However, if you want to specify
NN you need to give a value for the other parameters as well.
首先需要将OPC水的原子类型加入需要配合使用的力场文件中:
1 2 3
;To use this opc.itp, you must also add below lines into [ atomtypes ] of ffnonbonded.itp under AMBER forcefield ;OW_opc 8 15.9994 0.0000 A 0.316655 0.89036 ;HW_opc 1 1.0080 0.0000 A 0.0 0.0
[ atoms ] ; id at type res nr res name at name cg nr charge mass 1 OW_opc 1 SOL OW 1 0 16.00000 2 HW_opc 1 SOL HW1 1 0.6791 1.00800 3 HW_opc 1 SOL HW2 1 0.6791 1.00800 4 MW 1 SOL MW 1 -1.3582 0.00000
#else ;OPC paper didn't define force constant for flexible mode, so the FC values are set to TIP4P-Ew ones [ bonds ] ; i j funct length force.c. 1 2 1 0.08724 502416.0 0.08724 502416.0 1 3 1 0.08724 502416.0 0.08724 502416.0 [ angles ] ; i j k funct angle force.c. 2 1 3 1 103.6 628.02 103.6 628.02
#endif
[ virtual_sites3 ] ; Vsite from funct a b 4 1 2 3 1 0.14772952 0.14772952 ; ; a = b = distance(O-MW) / [ cos (angle(MW-O-H)) * distance(OH) ] /2 ; = 0.01594/(cos(103.6/2/180*3.141592653589793)*0.08724)/2
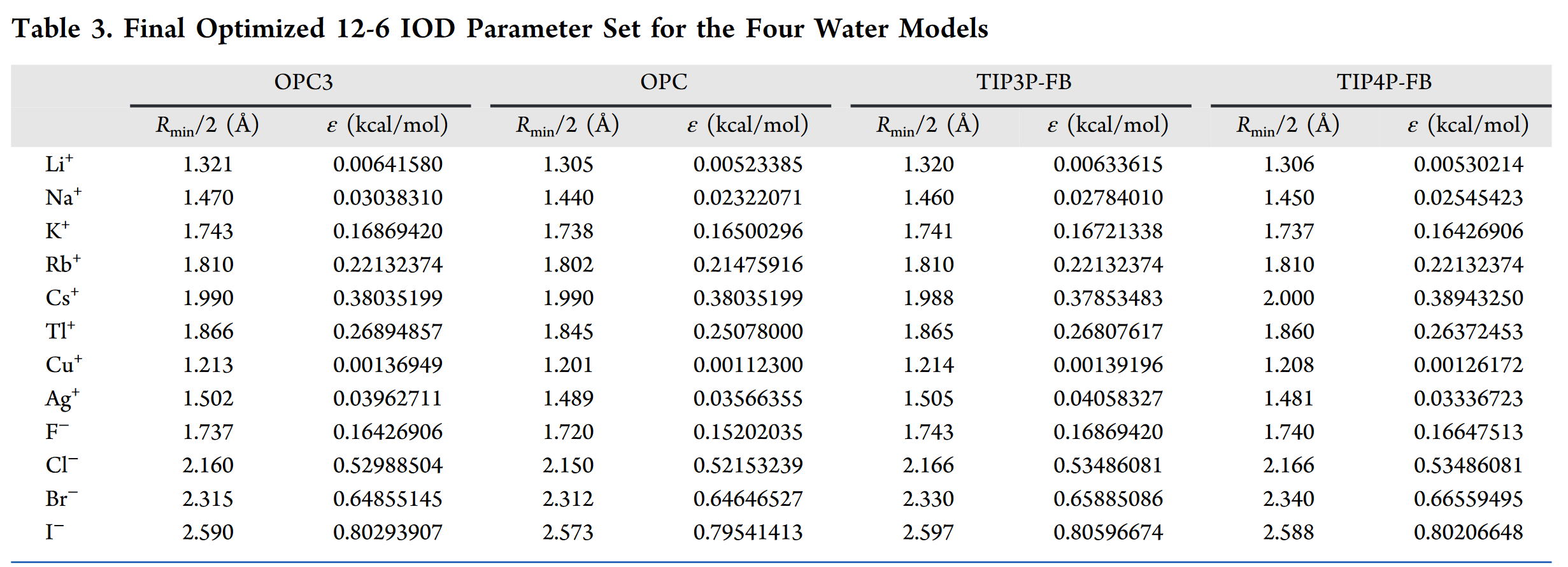
Li Li 6.94 0.00000 A 0.11769 0.02683 Na Na 22.99 0.00000 A 0.13096 0.12706 K K 39.10 0.00000 A 0.15528 0.70548 Rb Rb 85.47 0.00000 A 0.16125 0.92558 Cs Cs 132.91 0.00000 A 0.17729 1.59063 Tl Tl 204.38 0.00000 A 0.16624 1.12474 Cu Cu 63.546 0.00000 A 0.10807 0.00573 Ag Ag 107.87 0.00000 A 0.13381 0.16572 F F 19.00 0.00000 A 0.15475 0.68697 Cl Cl 35.45 0.00000 A 0.19243 2.21598 Br Br 79.90 0.00000 A 0.20624 2.71224 I I 126.9 0.00000 A 0.23074 3.35789
; Include water topology #include "opc.itp" #ifdef POSRES_WATER ; Position restraint for each water oxygen [ position_restraints ] ; i funct fcx fcy fcz 1 1 1000 1000 1000 #endif ; Include topology for ions #include "opc_ions.itp"
; LINES STARTING WITH ';' ARE COMMENTS title = Minimization ; Title of run
; Parameters describing what to do, when to stop and what to save integrator = steep ; Algorithm (steep = steepest descent minimization) emtol = 1000.0 ; Stop minimization when the maximum force < 10.0 kJ/mol emstep = 0.01 ; Energy step size nsteps = 50000 ; Maximum number of (minimization) steps to perform
; Parameters describing how to find the neighbors of each atom and how to calculate the interactions nstlist = 1 ; Frequency to update the neighbor list and long range forces cutoff-scheme = Verlet ns_type = grid ; Method to determine neighbor list (simple, grid) rlist = 1.0 ; Cut-off for making neighbor list (short range forces) coulombtype = cutoff ; Treatment of long range electrostatic interactions rcoulomb = 1.0 ; long range electrostatic cut-off rvdw = 1.0 ; long range Van der Waals cut-off pbc = xyz ; Periodic Boundary Conditions
; minim.mdp - used as input into grompp to generate em.tpr title = Minimization integrator = steep ; Algorithm (steep = steepest descent minimization) emtol = 500.0 ; Stop minimization when the maximum force < 50.0 kJ/mol/nm emstep = 0.01 ; Energy step size nsteps = 50000 ; Maximum number of (minimization) steps to perform comm-mode = Linear ; remove translation of center of mass ; Parameters describing how to find the neighbors of each atom and how to calculate the interactions cutoff-scheme = Verlet ns_type = grid ; Method to determine neighbor list (simple, grid) coulombtype = PME ; Treatment of long range electrostatic interaction rcoulomb = 1.2 ; Short-range electrostatic cut-off rvdw = 1.2 ; Short-range Van der Waals cut-off pbc = xyz ; Periodic Boundary Conditions (yes/no)
Izadi, S.; Anandakrishnan, R.; Onufriev, A. V. Building Water
Models: A Different Approach. J. Phys. Chem. Lett.2014, 5 (21), 3863–3871.
https://doi.org/10.1021/jz501780a.
Sengupta, A.; Li, Z.; Song, L. F.; Li, P.; Merz, K. M.
Parameterization of Monovalent Ions for the OPC3, OPC, TIP3P-FB, and
TIP4P-FB Water Models. J. Chem. Inf. Model.2021, 61 (2), 869–880.
https://doi.org/10.1021/acs.jcim.0c01390.